Home
![]()
![]()
Welcome to the documentation for the nf-illumina2lineage pipeline.
Overview¶
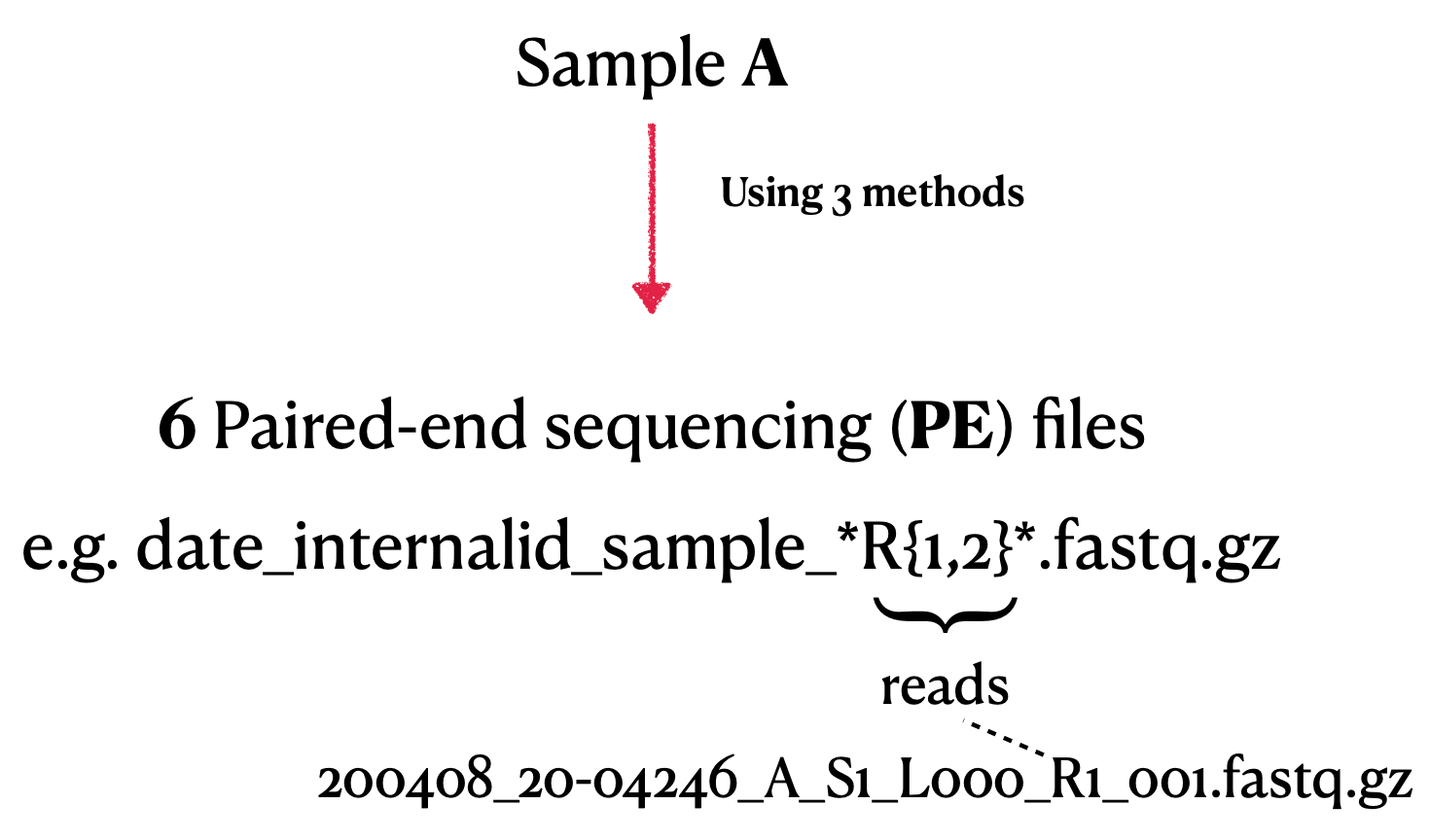
This repository provides a complete pipeline for assembling and analyzing the genome of SARS-CoV-2 using Illumina paired-end sequencing data. It includes steps for quality control, mapping, variant calling, primer clipping, consensus sequence generation, lineage annotation, and phylogenetic analysis.
Key Features¶
- Automated environment setup using
pixi - Comprehensive quality control with
fastqcandfastp - Mapping and visualization using
minimap2,samtools, andIGV - Primer sequence clipping for clean alignments
- Variant calling with
freebayes - Consensus sequence generation and lineage assignment with
pangolin - Phylogenetic analysis and multiple sequence alignment with
mafftandiqtree - Clear documentation and modular structure
System Requirements¶
- Operating System: Linux (tested on Fedora 38)
- Processor: Intel i5 or higher, with multithreading support
- Memory: Minimum 8 GB
Dependencies¶
The pipeline requires the following tools, managed via mamba:
- QC:
fastqc,fastp,multiqc - Mapping:
minimap2,samtools,bamclipper - Variant Calling:
freebayes,vcftools,bcftools - Sequence Analysis:
mafft,iqtree,pangolin - Visualization:
IGV
Input Data¶
- Illumina paired-end sequencing data
- SARS-CoV-2 reference genome (NCBI accession: NC_045512.2)
Output¶
- Quality control reports (
.html,.json) - Aligned sequences in BAM and VCF formats
- Consensus sequences in FASTA format
- Lineage annotations
- Phylogenetic trees and visualizations